Cisztás fibrózis
A CFTR gén mutációiA cisztás fibrózis (CF, cisztikus fibrózis, mukoviszcidózis) az egyik leggyakoribb veleszületett genetikai betegség, melyet a CFTR (cisztás fibrózis transzmembrán konduktancia regulátor) génben fellelhető eltérések (mutációk) okoznak.
Magyarországon minden 28. emberben a CFTR gén egyik kópiája hibás. Mára már több mint 1500 eltérést azonosítottak a CFTR génen belül. Kiemelendő a ΔF508del nevű mutáció, amely a betegek mintegy kétharmadánál felelős a kórkép kialakulásáért. A mutációk további egyharmada elszórtan lelhető fel a génben.
A CF gyakorisága indokolttá teszi a CFTR gén mutációs analízisét. Ez a vizsgálat Magyarországon egyelőre nem tartozik bele az újszülötteknél rutinszerűen elvégzett szűrésekbe, azonban több fejlett ország már jelenleg is felvette a rutin újszülöttkori szűrővizsgálatok közé.
A betegségben hiányzik, vagy nem működik jól a CFTR fehérje, ami az alábbi tüneteket okozza:
- A hasnyálmirigyben: emésztési problémák.
- A légzőrendszerben: krónikus, valamint rohamokban jelentkező köhögés, ismétlődő és krónikus tüdőgyulladás. Légzési problémák lépnek fel, sűrű váladék rakódik le a légutakban, emiatt a tüdő nem kap elegendő oxigént, így fokozatosan elhal.
- A mellékherékben: a mellékherék csatornáinak elzáródása miatt a férfi betegek 95%-a steril.
- A besűrűsödött nyák elzárja a külső elválasztású mirigyek kivezető csöveit, ami megváltozott nyáktermelésében mutatkozik meg.
- A csecsemőknél a meconium ileus, azaz a belek tartalmának besűrűsödése miatt bekövetkező elzáródás hívja fel a figyelmet a betegségre.
- Fehérjehiány
- A zsíroldékony vitaminok (A, D, E, K) hiányának jelei.
- Jellemző szinte az összes mirigy gyulladásos elváltozása.
A laborunkban kifejlesztett CFTR Diagnosztikai Kit lehetővé teszi a teljes gén analízisét, azaz az összes mutáció kimutatására alkalmas.
Cisztás fibrózisban szenvedő kisgyermek
A mukoviszcidózisos csecsemők tüdeje születéskor még normális, a tüdő és légutak nyálkahártyájának mirigyei azonban folyamatosan kórosan sűrű, nyákos váladékot termelnek, amely eltömi a léghólyagocskákat is, a tüdő légzőfelülete jelentősen csökken. A sűrű váladék nem tud természetes módon távozni, ezért a beteg kisgyermekek hosszas, kínzó köhögés kíséretében próbálnak tőle megszabadulni. Kezdetben a köhögés száraz, erős, szaggatott, később "felszakadozó", mintha oldódna. A sűrű váladék kedvez a különböző légúti fertőzések kórokozói megtelepedésének. Leggyakrabban a Staphylococcus aureus baktérium és a Pseudomonas fajok okoznak fertőzéseket. A legveszélyesebb a Pseudomonas aeruginosa, mely halálhoz vezető fertőzést okozhat.
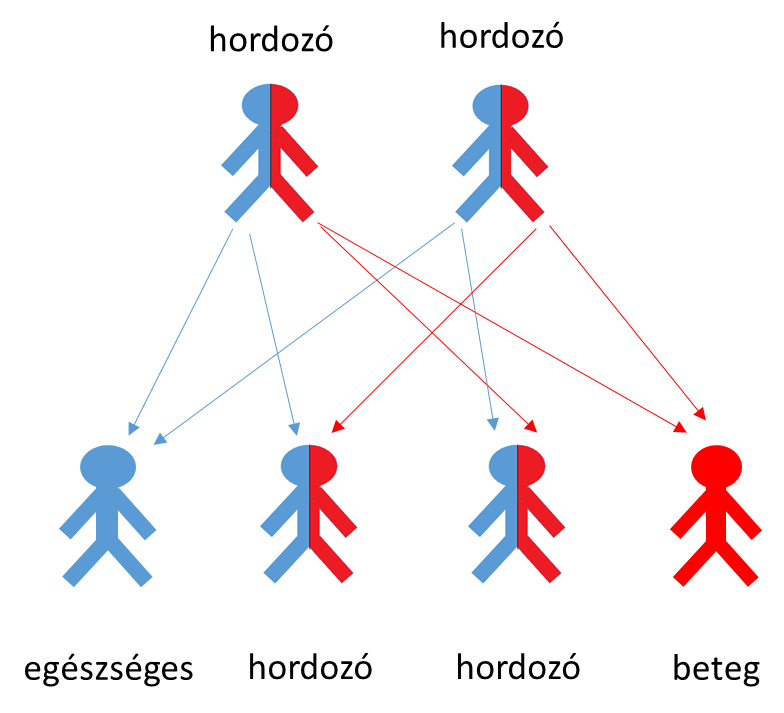
A betegség öröklődése a szülőkről a gyerekekre
Ha adott esetben mindkét szülő hordozza a mutációt a CFTR génben, akkor 25% annak a valószínűsége, hogy gyermekük beteg lesz.
Gyakran ismételt kérdések
A leggyakoribb öröklődő betegség, a cisztás fibrózis hátterében már 1989 óta tudjuk, hogy a CFTR gén meghibásodása áll. A betegséget okozó génvariánsok száma azonban a mai napig is folyamatosan bővül, ahogyan a különböző súlyosságú és lefolyású páciensek genetikai vizsgálatával a génnek illetve az őt kódoló fehérjének újabb és újabb defektusára kerül fény.

New and emerging targeted therapies for cystic fibrosis
Bradley S Quon, clinician-scientist1 and Steven M Rowe, director2
Ezek az eredmények alapozzák meg ennek a heterogén kórképnek a különböző variánsaira alkalmazható célzottan ható molekulák kiválasztását.
Cystic fibrosis genetics: from molecular understanding to clinical application.
Cutting GR. Nat Rev Genet. 2015 Jan;16(1):45-56. doi: 10.1038/nrg3849. Epub 2014 Nov 18. Review.